(Link to the first editorial: https://wp.me/pbW3AH-1DO -Link to the third editorial of this series: https://wp.me/pbW3AH-1Tx)
Based on the logic enlightened by scientific publications that have accumulated over decades, this series of editorials aims to offer the reader a path to identifying the etiology (cause) of autism—a condition whose annual cost, including direct and indirect expenses, could exceed 460 billion dollars in the U.S. by 2025 (https://pubmed.ncbi.nlm.nih.gov/26183723/).
The previous editorial brought together clinical and scientific data that identify the inflammatory nature of the pathological process causing autistic behavior. It is, therefore, unquestionably excluded (through a simple laboratory test, such as measuring the circulating levels of the enzyme Neuronal Specific Enolase – NSE) the hypothesis that autistic children would be just “neuroatypical” individuals. This term mistakenly regards them as “different” and not as carriers of deficits of an organic nature (carriers of a physical illness). In this sense, it is worth remembering that up to 60% of autistic people have a cognitive impairment (https://www.cdc.gov/mmwr/volumes/72/ss/ss7202a1.htm).
The release of NSE into circulation by damaged neuronal cells is eloquent enough to define autism as a neurological disease (with pathologic features consistent with chronic encephalitis). Persistent denial of this fact only hinders the development and timely application of early and effective treatment—a right of the affected children and their parents. With autism acknowledged as a neurological disorder, pinpointing its fundamental cause would enable preventive strategies and stop the ongoing autism epidemic, which is in the best interest of society (including the medical community). As will be seen in the next Editorial, the autism epidemic can no longer be regarded as merely apparent or as a result of “better diagnostics and awareness” (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6242891/), which led to the coining of the expression “Autism Spectrum Disorders” (ASD) instead of “autism”. In fact, how can one possibly believe that a simple pre-existing behavioral disorder, whose prevalence allegedly is not increasing worldwide (just being better “perceived” or “detected”), could be expected to generate an annual cost of more than 460 billion dollars by 2025 in the USA alone (https://pubmed.ncbi.nlm.nih.gov/26183723/)?
An inflammatory process that affects the brain (encephalitis) can, for example, be caused by viral or bacterial infections or even by the aggression of the immune system (autoimmune encephalitis). In addition, there is a fundamental fact regarding autism (which will be reviewed in subsequent editorials on this site): the chronic encephalitis characterizing this condition can be caused by the presence of aluminum in the nervous tissue (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10180736/), triggering an autoimmune process directed against neural proteins (antigens).
This Second Editorial focuses on the chronic autoimmune aggression present in individuals with autism (chronic encephalitis). The avalanche of scientific data demonstrating the presence of an autoimmune process in this condition contrasts with the view that autism is a simple behavioral disorder (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5373490/). As we will see in subsequent editorials, in line with the renowned “Principle of Parsimony,” which inspired modern statistics, one must identify the primary triggering factor of this autoimmune aggression, around which several secondary factors contributing to its progression and maintenance gravitate. That principle is used, for example, in daily medical practice to establish a single diagnosis (the most likely one), as opposed to the improbability of multiple diagnoses co-occurring in a single patient. When identifying the diagnosis of a disease, i.e., the single cause of a constellation of clinical and laboratory manifestations, it is essential to pursue the most straightforward explanation (ideally the only one) for all manifestations.
Thus, just as it is improbable that a patient will present with two diseases that begin simultaneously without any pathophysiological relationship between them, it is also unlikely that the pieces of the pathophysiological puzzle of autism will not fit together perfectly around an overriding causal factor.
Likewise, all the discoveries reported in autism, including its condition of chronic encephalitis, the autoimmune features, the predisposing genetic factors, and mainly, the exponential growth of its incidence and prevalence that has occurred in the last 40 to 50 years, must be grouped and arranged around a primary cause, much like the process of assembling the pieces of a jigsaw puzzle. Again, it is emphasized here that without identifying this primary cause, there is no way to stop the pathological (inflammatory, autoimmune, neurodegenerative) process that characterizes autism or to implement its prevention.
The statement that “autism has multiple causes that occur in various combinations” or that it is “a highly complex and heterogeneous biological disorder” (https://www.biologicalpsychiatryjournal.com/article/S0006-3223(16)32739-1/) contradicts the Principle of Parsimony (or “Occam’s Razor”). Such statements only serve to evade the search for the primary cause of this humanitarian, social, and economic tragedy while its prevalence continues to rise rapidly without preventive measures to counter it. On the contrary, in this series of editorials, we seek to equate the diversity of pathophysiological findings related to autism with the various clinical and laboratory manifestations found in a patient to identify a single primary cause—comparatively, a single diagnosis as opposed to multiple diagnoses.
On the other hand, the statement that “a person with autism is born with autism” (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5501015/) disregards the currently undisputed fact that many children develop the first manifestations of autism after the first year of life, following a perfectly normal initial psychomotor development (“regressive autism” – which was speculatively denied for a long time) (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4949854/).
However, regressive autism does exist (https://goldencaretherapy.com/regressive-autism – 2021):
“Regressive autism, also known as late-onset autism, involves a period of typical development followed by a loss of previously acquired skills or a noticeable decline in social and communication abilities. This regression usually occurs between 15 and 30 months of age and can be sudden or gradual.”
The following example was taken from a text written a few years ago. At that time (2017), the author was trying to argue against the reality of the existence of cases of regressive autism (https://www.thetransmitter.org/spectrum/rethinking-regression-autism/):
“…a talkative, curious 2-year-old suddenly withdraws. He grows indifferent to the sound of his name. He begins to speak less than before or stops entirely. He turns from playing with people to playing with things, from exploring many objects and activities to obsessing over a few. He loses many of the skills he had mastered and starts to rock, spin, walk on his toes or flap his hands. It’s often at this point that his terrified parents seek answers from experts.”
…and parents do not usually find assertive answers consistent with scientific logic—only speculations. This is due to the lack of an integrated and rational presentation of the pathophysiological features already documented in various studies. Such a gap is precisely the objective that inspired these consecutive editorials, which should also explain the existence of regressive autism.
Contrary to attempts to deny its existence, the reality of regressive autism has been recognized for many years in scientific publications, where the authors point it out as “intriguing” (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4949854/), thus acknowledging that they do not know how to explain it:
“The occurrence of developmental regression in autism spectrum disorder (ASD) is one of the most puzzling phenomena of this disorder.”
“To date, the causes of regression in autism are unknown.”
In this editorial, we review the direct impact of this autoimmune aggression on the access of a vitally important nutrient to the central nervous system (CNS): vitamin B9 (or “folate”: methylfolate and folinic acid—the latter also called leucovorin). Specific genetic polymorphisms (genes with a structure different from the standard conformation) make a significant fraction of the child population particularly susceptible to a reduction in the supply of folates to the CNS.
Autoimmune aggression towards the brain in autistic children is confirmed through repeated publications beginning in the 1980s and subsequently reviewed by several authors (https://www.sciencedirect.com/science/article/pii/S1750946720300581; https://www.frontiersin.org/journals/cellular-neuroscience/articles/10.3389/fncel.2018.00405/full#B90; https://psychiatryonline.org/doi/10.1176/appi.focus.24022004; https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8955336/; https://www.nature.com/articles/npp2016158; https://psychiatryonline.org/doi/10.1176/appi.focus.24022004;
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6242891/; https://pubmed.ncbi.nlm.nih.gov/15223250/;
The evidence suggesting that chronic encephalitis in autistic individuals involves an autoimmune attack against brain tissue can be arranged as follows:
(I) Generation of autoantibodies targeting neural antigens;
(II) Generation of autoantibodies not specifically targeting neural antigens;
(III) Correlation between circulating autoantibody levels and the severity of autism;
(IV) Presence of lymphocytes infiltrating the brain of individuals with autism;
(V) Th17 immune response (induced by interleukin 17-producing helper lymphocytes, typical in autoimmune conditions) has been observed in autistic individuals;
(VI) Mitigation of autistic symptoms through corticosteroid or immunoglobulin therapy;
(VII) Occurrence of autoimmune or immune-mediated disorders as comorbidities in autism;
(VIII) Elevated prevalence of autoimmune diseases among consanguineous relatives (a family history of autoimmune disorders increases autism risk);
(IX) Similar to classic autoimmune diseases, the benefits of cholecalciferol administration are also observed in autism;
(X) Evidence pieces show genetic resistance to the immunoregulatory effects of cholecalciferol (or “vitamin” D, which inhibits Th17 activity) in autism
Below is the presentation of each group of publications.
(I) Generation of autoantibodies targeting neural antigens.
The immune system of individuals with autism generates a wide variety of autoantibodies targeting neural antigens (https://www.frontiersin.org/journals/cellular-neuroscience/articles/10.3389/fncel.2018.00405/full#B90; https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6952169/), with reactivity directed against various regions of the CNS and different populations of neurons. Together with the other published data listed below, the production of autoantibodies cannot be considered merely an epiphenomenon of the process causing autism; rather, it should be regarded as a cardinal factor in the inflammatory process harmful to the CNS (an epiphenomenon would be a phenomenon associated with a lesional process without a causal relationship to it).
According to the Principle of Parsimony, the primary cause of autism must explain both the broad repertoire of autoantibodies produced and the variety of neural regions and cells affected. As mentioned in the introduction to this Editorial, the primary cause around which aggravating factors gravitate will be identified in a future Editorial, applying the causality criteria established by Austin Bradford Hill in 1965 and widely recognized since then.
Singh et al. first identified the presence of autoantibodies directed against CNS proteins in autistic individuals in 1988. They found antibodies against neuron axon filament protein (NAFP) in the blood of 10 out of 15 children with autism (https://pubmed.ncbi.nlm.nih.gov/3144935/). Anti-myelin basic protein (MBP) antibodies were identified in individuals with ASD in 1993 (https://pubmed.ncbi.nlm.nih.gov/7682457/)—a finding that was replicated in 1998 (https://pubmed.ncbi.nlm.nih.gov/9756729/) and again in 2006 (https://pubmed.ncbi.nlm.nih.gov/16181614/). The presence of anti-NAFP antibodies was also confirmed in 1998 (https://pubmed.ncbi.nlm.nih.gov/9756729/). In 2013, significantly elevated levels of anti-MBP antibodies were confirmed compared to healthy controls (https://pubmed.ncbi.nlm.nih.gov/23726766/). The same study demonstrated significantly elevated levels of anti-myelin-associated glycoprotein (“anti-MAG”) antibodies compared to healthy controls. The same study also demonstrated the link between these autoantibodies and autism severity:
“Patients with severe autism had significantly higher serum anti-MBP and anti-MAG auto-antibodies than children with mild to moderate autism, P = 0.047 and P < 0.001, respectively (Tables 1 and 2).”
Other studies have demonstrated a significant increase in the incidence of anti-NAFP and anti-glial fibrillary acidic protein (GFAP) in autistic individuals, but not in individuals with mental retardation (https://www.sciencedirect.com/science/article/abs/pii/S0887899497000453).
More recent studies have found autoantibodies targeting regions of the prefrontal cortex, caudate, putamen, cerebellum, and cingulate gyrus of the brain (https://pubmed.ncbi.nlm.nih.gov/16842863/) and hypothalamus (https://pubmed.ncbi.nlm.nih.gov/17804536/) in children with ASD.
As the authors of this latest study conclude:
“While the potential role of these autoantibodies in autism is currently unknown, their presence suggests a loss of self-tolerance to one or more neural antigens during early childhood.”
Similarly, in another study published in 2009 (https://pubmed.ncbi.nlm.nih.gov/18706993/), researchers found that 21% of plasma samples from children with ASD were highly immunoreactive against primate Golgi neurons. These inhibitory interneurons are located in the granular layer of the cerebellum. They use the neurotransmitter gamma-aminobutyric acid (GABA) to modulate excitatory synapses, enabling a balance between excitation and inhibition. In contrast, this immunoreactivity was not observed when plasma obtained from age-matched, typically developing controls was used. A later study found that autoreactivity also targeted other GABAergic interneurons distributed throughout the neocortex and in many subcortical regions, including the superficial layers of the cortex (https://pubmed.ncbi.nlm.nih.gov/21521495/). Other authors found that immunoreactivity directed at Golgi neurons and other interneurons correlates with the severity of behavioral and emotional changes in autistic children (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3313674/; https://pubmed.ncbi.nlm.nih.gov/21420487/;https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3039058/; https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4111628/; https://pubmed.ncbi.nlm.nih.gov/22226851). In line with the results of these studies, alterations in the structure of the cerebellum and in the composition of the cerebellar neuron population are among the most consistent abnormalities in autism (https://pubmed.ncbi.nlm.nih.gov/11468308/).
Contrary to what was previously thought, immune system cells routinely penetrate the CNS, even in the absence of inflammation, to perform immunological surveillance. Local presentation of antigens to produce antibodies and the proliferation of lymphocyte clones occur even in the noninflamed nervous system (https://www.nature.com/articles/nn.3161). In neuroinflammation, the barriers that separate the brain from circulation are disrupted, thus increasing the transit of cells and macromolecules between these compartments. (https://www.mdpi.com/1422-0067/24/16/12699). The penetration of autoantibodies directed against GABAergic neurons may, therefore, reduce the number or activity of these inhibitory cells. This may contribute to the imbalance between excitation and inhibition—an imbalance that has long been suggested as a determinant of the disturbance of sensory, memory, social, and emotional systems found in autism (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6748642/).
Glial cells (which help shield, support, and nourish neurons) have long been known to multiply (through a process called mitosis). Contrary to older concepts, the generation of new neurons has also been demonstrated in the adult human CNS for over a quarter of a century, although not through mitosis. Newly formed neurons arise from the multiplication of cells located around the cavities and channels through which cerebrospinal fluid circulates—the “subependymal” layer. The term “ependyma” is used to indicate the layer of cells that lines the internal cavities of the brain (ventricles), while the “subependymal” region denotes the area adjacent to the ventricles. The cells in this region can be classified into two types: neural stem cells and neural progenitor cells, the latter of which are derived from neural stem cells. When they multiply, both lead to the formation of new neurons (neurogenesis) and two types of glial cells: astrocytes and oligodendrocytes, which constitute the “macroglia”. They participate in the formation and maturation of the neural system, not only in the embryonic and fetal phases but also after birth, and the production of new neurons continue even in elderly individuals. These newly formed neurons (“neuroblasts”) are capable of migrating from the subependymal region to various regions of the CNS, supporting the neural cell population and function of these regions (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6782600/; https://www.jneurosci.org/content/22/3/612.short; https://www.nature.com/articles/nm1198_1313; https://www.sciencedirect.com/science/article/pii/S2214854X20300133).
Neurogenesis is probably involved in the processes of skill acquisition in childhood, such as speech (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3844860/). On the other hand, the normal development of the nervous system during the postnatal period requires the elimination of an excess of connections (synapses) between neurons through a process called “synaptic pruning” (https://www.science.org/doi/abs/10.1126/science.1202529). This process is inhibited in the brains of individuals with autism (https://www.nature.com/articles/mp2016103).
A high serum level of autoantibodies generated against human neural progenitor cells has been identified in patients with autism (https://pubmed.ncbi.nlm.nih.gov/23838310/). On the other hand, serum from autism patients suppresses the differentiation and maturation of neural progenitor cells in culture, demonstrating an autoimmune event that may be one of the mechanisms of neurodevelopmental impairment in this condition, involving the inhibition of neurogenesis (https://pubmed.ncbi.nlm.nih.gov/19526302/; https://pubmed.ncbi.nlm.nih.gov/19526302/; https://pubmed.ncbi.nlm.nih.gov/23838310/).
On the other hand, excessive microglial activation, as part of the mechanism involved in the inflammation that affects the autistic brain (https://www.sciencedirect.com/science/article/abs/pii/S0022395620308785), can alter the execution of the neural pruning process by these cells. This may lead to an excess of excitatory synapses to the detriment of inhibitory synapses (https://www.sciencedirect.com/science/article/abs/pii/S0168010215001625), thus contributing, also through this mechanism, to a hyperexcitation potentially underlying the hyperactivity found in autistic individuals.
Contributing to this imbalance is the production of autoantibodies directed against serotonin receptors in the autistic brain (https://pubmed.ncbi.nlm.nih.gov/2578670/; https://pubmed.ncbi.nlm.nih.gov/1375597/). Serotonin is a neurotransmitter of fundamental importance for emotional behavior, and the impairment of serotonergic activity can alter attention and emotional reactions to external stimuli (https://www.sciencedirect.com/science/article/abs/pii/S1569733910700904), as seen in autistic behavior (https://www.sciencedirect.com/science/article/abs/pii/S0890856713003080). The antibodies directed against the serotonin receptor in an autistic child reached values of 600 and 980 fmol/ml of serum in two samples collected one month apart. These levels are much higher than the levels of antibodies directed against the nicotinic receptors of the neurotransmitter acetylcholine, found in a disease classically recognized as autoimmune (myasthenia gravis), in which serum autoantibody titers reach maximum values of 45 fmol/ml, resulting in a reduction in skeletal muscle strength (https://pubmed.ncbi.nlm.nih.gov/2578670/).
Among the autoantibodies directed against brain cells found in the serum of autistic children are also antibodies against myelin-associated basic glycoprotein (anti-MAG) (https://pubmed.ncbi.nlm.nih.gov/22898564/; https://pubmed.ncbi.nlm.nih.gov/23726766/), anti-myelin basic protein (anti-MBP) (https://pubmed.ncbi.nlm.nih.gov/23726766/), and anti-ganglioside M1 antibodies (the most abundant glycosphingolipid component of neuronal membranes). The highest levels of these autoantibodies are found in more severe cases of autism (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3104945/).
Other specific brain autoantibodies, such as anti-myelin basic protein antibodies (https://pubmed.ncbi.nlm.nih.gov/15223250/), anti-myelin-associated glycoprotein antibodies, and anti-ganglioside M1 antibodies (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3104945/), have been observed in autistic children.
(II) Generation of autoantibodies not specifically targeting neural antigens.
The antinuclear antibody (ANA) is composed of antibodies directed against structures (antigens) located in the nucleus and cytoplasm of cells, such as proteins, nucleic acids (DNA, RNA), and protein-nucleic acid complexes. For decades, its presence in the circulation has been considered fundamental for diagnosing autoimmune diseases (https://ard.bmj.com/content/73/1/17.short; https://www.sciencedirect.com/science/article/abs/pii/S2173574310700496). Its production by the cells of an individual’s immune system (also in children) indicates that an autoimmune disease may be present or developing, such as systemic lupus erythematosus (SLE), scleroderma (localized or systemic), mixed connective tissue disease (with mixed features of SLE, and polymyositis), rheumatoid arthritis, juvenile rheumatoid arthritis, Sjögren’s syndrome, polymyositis and dermatomyositis (https://my.clevelandclinic.org/health/diagnostics/14897-antinuclear-antibody-test-in-children; https://www.researchgate.net/publication/333705452_Pattern_and_Frequency_of_Anti-nuclear_Antibody_Positivity_in_Paediatric_Rheumatic_Diseases).
As would be expected in the case of the participation of autoimmune mechanisms in autism, a positive result on the ANA test has been found in autistic children, associated with the severity of autistic manifestations and the presence of electroencephalographic alterations (https://pubmed.ncbi.nlm.nih.gov/19135624/).
Antiphospholipid antibodies are autoantibodies that target proteins bound to phospholipids (fundamental lipid structures in cell membranes) (https://pt.khanacademy.org/science/ap-biology/cell-structure-and-function/plasma-membranes/a/structure-of-the-plasma-membrane#:~:text=v%C3%AAm%20dos%20carboidratos.-,Fosfolip%C3%ADdios,t%C3%AAm%20regi%C3%B5es%20hidrof%C3%ADlicas%20e%20hidrof%C3%B3bicas). The presence of these antibodies leads to antiphospholipid antibody syndrome (APS—a multisystem autoimmune disease—https://pubmed.ncbi.nlm.nih.gov/36849186/). The result is an increased risk of thrombotic events, pregnancy morbidity, and several other autoimmune and inflammatory complications (https://pubmed.ncbi.nlm.nih.gov/36849186/). Although it was initially described in the context of lupus (SLE), APS is also found to be dissociated from SLE with similar frequency (https://pubmed.ncbi.nlm.nih.gov/36849186/). In addition to SLE, diseases such as thrombocytopenia, hemolytic anemia, heart valve disease, pulmonary hypertension, microangiopathic nephropathy, skin ulcers, livedo reticularis, refractory migraine, cognitive dysfunction, and atherosclerosis are associated with APS (https://www.ncbi.nlm.nih.gov/books/NBK459442/). Supporting the involvement of autoimmune mechanisms in autism, elevated levels of antiphospholipid autoantibodies (anti-cardiolipin, anti-β2-glycoprotein 1, and anti-phosphoserine) have been found in individuals with autism and are associated with the severity of behavioral changes (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3794552/).
Anti-endothelial cell antibodies are a heterogeneous group of antibodies directed against the cells lining blood vessels. Since their discovery in the 1970s, these autoantibodies have been identified in several conditions characterized by vascular inflammation, including SLE, APS, systemic vasculitis, rheumatoid arthritis, systemic scleroderma, and organ transplant rejection (https://www.sciencedirect.com/science/article/abs/pii/S1568997216302798). Similar to what occurs with ANA and antiphospholipid antibodies, the presence of higher levels of circulating anti-endothelial cell antibodies has a direct relationship to the severity of behavioral changes in people with autism (https://www.sciencedirect.com/science/article/pii/S1726490115000969?via%3Dihub).
The detection of antimitochondrial antibodies (AMA) is used to diagnose the autoimmune disease called primary biliary cholangitis. It may also occur in other autoimmune diseases, such as Sjögren’s syndrome, systemic sclerosis (or scleroderma), polymyositis/dermatomyositis, juvenile idiopathic arthritis, SLE, and autoimmune hepatitis (https://link.springer.com/article/10.1007/s12016-021-08904-y; https://www.sciencedirect.com/science/article/pii/S2589909022000065: https://www.sciencedirect.com/science/article/abs/pii/S0889857X05702970). Anti-double-stranded DNA antibodies are considered highly specific markers of SLE (https://www.nature.com/articles/s41584-020-0480-7) and lupoid autoimmune hepatitis (https://www.nature.com/articles/s41584-021-00573-7). The presence of AMA (https://jneuroinflammation.biomedcentral.com/articles/10.1186/1742-2094-7-80; https://pubmed.ncbi.nlm.nih.gov/24837704/), anti-double-stranded DNA antibodies and ANA (https://pubmed.ncbi.nlm.nih.gov/24837704/) and anti-nucleosome antibodies (https://pubmed.ncbi.nlm.nih.gov/24708718/) have also been documented in the serum of autistic individuals (nucleosomes are structural units that form chromosomes and are composed of two spirals of DNA wrapped around a protein disc consisting of four pairs of proteins called histones). The anti-nucleosome antibody test is considered highly sensitive and specific for the diagnosis of SLE (https://pubmed.ncbi.nlm.nih.gov/20374326/). This indicates that the autoimmunity found in autism, which is part of the inflammatory process described in the first editorial of this series, may not only be organ-specific in many cases. In other words, it may not be solely directed against the brain or CNS of the autistic individual but rather a condition in which the immune system attacks other organs or systems.
Again, following the Principle of Parsimony, according to which the simplest and most comprehensive explanation should be considered the most likely for any phenomenon (including any disease), the search for the primary determining cause of autism must necessarily identify as causal a factor that can explain not only the aggression against the CNS but also the autoimmune aggression directed at other organs and systems.
(III) Correlation between circulating autoantibody levels and the severity of autism
Autoantibody levels have been identified as markers of the activity and severity of autoimmune diseases (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC209428/).
Corroborating the participation of the autoimmune phenomenon in the pathophysiology of autism, this correlation (direct relationship) has been documented in autistic children (https://www.sciencedirect.com/science/article/pii/S1726490115000969?via%3Dihub; https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3794552/; https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3039058/; https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3104945/; https://pubmed.ncbi.nlm.nih.gov/22226851; https://pubmed.ncbi.nlm.nih.gov/19135624/).
(IV) Presence of lymphocytes infiltrating the brain of individuals with autism
Lymphocyte accumulation is present in tissues and organs affected by autoimmune diseases. In association with inflammation and the production of autoantibodies, this finding (found in biopsies or autopsies under the microscope) is considered the hallmark of autoimmune diseases (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2832720/; https://pathology.jhu.edu/autoimmune/damage).
Similarly, lymphocytic infiltrates have been demonstrated in the brains of individuals with autism (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7210715/). Together with the evidence of an active brain inflammatory process (chronic encephalitis: first Editorial in this series) and the repertoire of antibodies produced against brain tissue listed here, the presence of autoreactive lymphocytes infiltrating the nervous tissue of autistic individuals completes the characterization of the triad classically considered the hallmark of autoimmune aggression involved in the pathophysiological mechanism of autism.
On the other hand, the discovery of active lymphatic vessels connecting the CNS to the lymphatic system (https://www.nature.com/articles/npp2016158; https://pubmed.ncbi.nlm.nih.gov/26077718/) indicates a possible communication channel through which autoimmune aggression may act in the autistic brain and in other autoimmune neurological diseases.
(V) The Th17 immune response (induced by interleukin 17-producing helper lymphocytes, typical in autoimmune conditions – https://link.springer.com/article/10.1007/s00281-019-00733-8) has been observed in autistic individuals (https://link.springer.com/article/10.1186/s13229-021-00472-4).
(VI) Mitigation of autistic symptoms through corticosteroid or immunoglobulin therapy
One of the most classic characteristics of autoimmune diseases is the vigorous response to the therapeutic use of drugs considered to be the most potent immunosuppressants and anti-inflammatories: glucocorticoids (https://pubmed.ncbi.nlm.nih.gov/11457656/). That also occurs in autoimmune diseases that affect the nervous system (https://pubmed.ncbi.nlm.nih.gov/11430999/), such as multiple sclerosis (https://pubmed.ncbi.nlm.nih.gov/23229226/). The core features of autism similarly respond to corticosteroid treatment (https://link.springer.com/article/10.1186/1471-2377-14-70; https://pubmed.ncbi.nlm.nih.gov/32168067/; https://www.scielo.br/j/jped/a/PBQNCqJ5L4cyqLXF5hyCxQy/?lang=en#), revealing an autoimmune mechanism involved in the pathophysiology of autism.
Likewise, autoimmune diseases may respond to immunoglobulin therapy when other therapeutic approaches fail (https://pubmed.ncbi.nlm.nih.gov/37062358/), as widely reported (https://ameripharmaspecialty.com/ivig-and-autoimmune-diseases/). As further evidence of the involvement of autoimmune mechanisms in autism, the same effect has been reported on the manifestations of this disorder (https://pubmed.ncbi.nlm.nih.gov/30097568/; https://www.mdpi.com/2075-4426/11/6/488).
(VII) Occurrence of autoimmune or immune-mediated disorders as comorbidities in autism
Allergies, asthma, atopic dermatitis, allergic rhinitis, urticaria, type 1 diabetes, inflammatory bowel disease (Crohn’s disease), and psoriasis are comorbidities of autism; that is, they are found in individuals with autism at a higher prevalence than in the general population or in individuals without autism (https://pubmed.ncbi.nlm.nih.gov/23726766/; https://jlb.onlinelibrary.wiley.com/doi/epdf/10.1189/jlb.1205707; https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10619695/; https://pubmed.ncbi.nlm.nih.gov/22511918/; https://www.sciencedirect.com/science/article/abs/pii/S1750946712001018; https://www.scirp.org/journal/paperinformation?paperid=78725; https://pubmed.ncbi.nlm.nih.gov/37939694/). The presence of atopic dermatitis is associated not only with a greater likelihood of autism but also with the greater severity of the autistic condition (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10619695/), suggesting a common causal factor for these two conditions.
(VIII) Elevated prevalence of autoimmune diseases among consanguineous relatives (a family history of autoimmune disorders increases autism risk)
This group of evidence demonstrates that children who have family members with autoimmune diseases, such as rheumatoid arthritis, systemic lupus erythematosus, celiac disease, ulcerative colitis, type 1 diabetes, hypothyroidism, Hashimoto’s thyroiditis, psoriasis, and rheumatic fever, are more likely to develop autism (https://pubmed.ncbi.nlm.nih.gov/19135624/; https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5373490/; https://pubmed.ncbi.nlm.nih.gov/25981892/; https://jamanetwork.com/journals/jamapediatrics/article-abstract/485932; https://pubmed.ncbi.nlm.nih.gov/14595086/; https://pubmed.ncbi.nlm.nih.gov/10385847/; https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3115699/; https://pubmed.ncbi.nlm.nih.gov/19581261/; https://pubmed.ncbi.nlm.nih.gov/16598435/; https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9025211/).
Children whose parents have autoimmune diseases may have a 50% higher risk of developing autism (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3115699/).
(IX) Similar to classic autoimmune diseases, the benefits of cholecalciferol administration are also observed in autism
“Vitamin” D (cholecalciferol, whose pre-active form is calcidiol or calcifediol and whose active form is calcitriol) has a structure similar to that of steroid hormones (estrogen, progesterone, testosterone, and cortisol). Like steroid hormones, it is derived from cholesterol, has receptors in the cell nucleus, and acts by modifying genetic activity—it modulates the activity of thousands of genes (https://www.mdpi.com/2073-4425/14/9/1691; https://www.sciencedirect.com/science/article/pii/S0039128X23000995). With nuclear receptors present in almost all, if not all, nucleated cells (https://www.sciencedirect.com/science/article/abs/pii/S0003986112001324; https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9325172/), as well as in cell membranes (https://pubmed.ncbi.nlm.nih.gov/17341182/), “vitamin” D (actually a hormone or a steroid hormone precursor) has pleiotropic (multiple) effects on the human organism. (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4045445/; https://www.sciencedirect.com/science/article/abs/pii/B9780323913867000064). Due to its multiple benefits for human health, to the fact that it is produced by exposing the skin to sunlight during the daytime hours that are mistakenly regarded as inappropriate (https://www.grassrootshealth.net/blog/shadow-can-tell-right-time-make-vitamin-d/), and to the confinement typical of modern urban life (https://link.springer.com/article/10.1186/s12889-017-4436-z; https://www.sciencedirect.com/science/article/abs/pii/B9780323913867000064), the deficiency of this pre-steroid hormone (“vitamin” D) has reached epidemic proportions (https://go.gale.com/ps/i.do?id=GALE%7CA592138121&sid=googleScholar&v=2.1&it=r&linkaccess=abs&issn=15228606&p=AONE&sw=w&userGroupName=anon%7E84477f50&aty=open-web-entry; https://academic.oup.com/nutritionreviews/article-abstract/81/10/1290/7071638?redirectedFrom=PDF&login=false; https://www.scielo.br/j/abem/a/78X5HHQSwzZtc435P9CsjCg/?lang=en). This deficiency has profound consequences for pre- and postnatal brain development that can extend throughout an individual’s lifespan (https://pubs.rsc.org/en/content/articlehtml/2023/fo/d3fo00166k).
Low vitamin D levels (masked by underestimated laboratory reference values) (https://www.grassrootshealth.net/wp-content/uploads/2017/05/dip_with_numbers_nmol_051317.pdf; https://www.mdpi.com/2072-6643/16/11/1666), “recommended” supplementation doses lower than those actually needed (https://www.mdpi.com/2227-9067/1/2/208), and genetic resistance to its effects (https://pubmed.ncbi.nlm.nih.gov/33897704/) have contributed to an increasing incidence of a wide variety of diseases, including multiple sclerosis, rheumatoid arthritis, inflammatory bowel diseases (Crohn’s disease, ulcerative colitis), celiac disease, uveitis, dermatological diseases, ankylosing spondylitis, fibromyalgia, diabetes, hypertension, tuberculosis, COVID-19, and cancer (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6032242/; https://link.springer.com/article/10.1007/s00223-019-00577-2; https://www.pnas.org/doi/abs/10.1073/pnas.1200072109; https://www.nature.com/articles/s41430-020-0661-0; https://apcz.umk.pl/QS/article/view/54077; https://www.sciencedirect.com/science/article/abs/pii/S0039625721001685; https://www.nature.com/articles/nrcardio.2009.135; https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2023.950465/full; https://pubmed.ncbi.nlm.nih.gov/11115787/; https://www.sciencedirect.com/science/article/abs/pii/S0009898114003921; https://www.sciencedirect.com/science/article/abs/pii/S0304395913005411).
As Wacker and Holick highlighted in their 2012 publication (https://www.mdpi.com/2072-6643/5/1/111):
“Vitamin D, the sunshine vitamin, has received a lot of attention recently as a result of a meteoric rise in the number of publications showing that vitamin D plays a crucial role in a plethora of physiological functions and associating vitamin D deficiency with many acute and chronic illnesses including disorders of calcium metabolism, autoimmune diseases, some cancers, type 2 diabetes mellitus, cardiovascular disease and infectious diseases.”
Patients with autoimmune diseases treated safely with high doses of vitamin D (https://www.mdpi.com/2072-6643/14/8/1575/review_report) may have polymorphisms (SNPs) affecting any combination of the nine genes that vitamin D requires to produce various biological effects, such as regulating the immune system (https://pubmed.ncbi.nlm.nih.gov/33897704/). Evidently, these genetic polymorphisms can cause resistance to the effects of vitamin D, impairing tolerance to autoantigens (https://pmc.ncbi.nlm.nih.gov/articles/PMC6712894/).
The fundamental roles of this steroid hormone (in the forms of calcidiol and calcitriol) in regulating and enhancing the immune system https://www.sciencedirect.com/science/article/abs/pii/S1471489210000378; https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2861286/; https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2023.1186635/full), as well as in the functions of the CNS (https://academic.oup.com/jbmrplus/article/5/1/e10419/7486306?login=false), are relevant to the present presentation.
As Eyles highlights in his article (https://www.mdpi.com/2072-6643/5/1/111):
“There is now also good evidence linking gestational and/or neonatal vitamin D deficiency with an increased risk of neurodevelopmental disorders, such as schizophrenia and autism, and adult vitamin D deficiency with certain degenerative conditions.”
Similar to its advantages in autoimmune diseases (https://www.nature.com/articles/ncprheum0855; https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2998156/), vitamin D supplementation also offers benefits for autism. In a recent umbrella review (2024)—a systematic analysis of various systematic reviews and meta-analyses on a specific topic—Jiang and colleagues (https://www.tandfonline.com/doi/full/10.2147/NDS.S470462#abstract) conclude:
“Based on rigorous analysis, we found that vitamin D deficiency early in life is a risk factor for the development of ASD and that vitamin D supplementation improves the core symptoms of ASD. Our study concludes that vitamin D supplementation is beneficial for individuals with autism, that vitamin D deficiency early in embryonic life increases the risk of ASD, and that our study supports the idea that prevention begins with vitamin D supplementation early in life.”
(X) Evidence pieces show genetic resistance to the immunoregulatory effects of cholecalciferol (or “vitamin” D, which inhibits Th17 activity) in autism
As is the case in autoimmune diseases in general, in which genetic polymorphisms related to vitamin D activity (https://www.cell.com/heliyon/fulltext/S2405-8440(24)03731-9) can cause resistance to its biological effects (including limiting its role in regulating the immune system) (https://pubmed.ncbi.nlm.nih.gov/33897704/), similar polymorphisms affecting the same genes have also been described in autism (https://www.mdpi.com/2076-3425/7/9/115; https://onlinelibrary.wiley.com/doi/abs/10.1002/aur.2279; https://www.sciencedirect.com/science/article/abs/pii/S0378111916303614; https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6207492/; https://www.sciencedirect.com/science/article/abs/pii/S037837821500119X).
Guerine and collaborators (https://onlinelibrary.wiley.com/doi/abs/10.1002/aur.2279) even observed a correlation between a specific polymorphism (SNP) in the vitamin D receptor (VDR) gene and the severity of autism:
“Finally, a strong gene-dose association of FokI (T) allele with both higher Childhood Autism Rating Scale score (Pc = 0.01) and, particularly, with hyperactivity behavior (Pc = 0.006) emerged in ASD children.”
These genetic polymorphisms, capable of causing resistance to the effects of vitamin D, indicate that administering higher doses (capable of compensating for the level of resistance), such as those used in psoriasis and vitiligo, can restore the immunomodulatory effects of vitamin D in autism as well. (https://pubmed.ncbi.nlm.nih.gov/24494059/).
In autism, autoimmunity damages the pathway that allows vitamin B9 to enter the Central Nervous System, leading to further harm to nerve cells.
Folate (or vitamin B9), whose natural forms are methylfolate (or methyltetrahydrofolate, the active form) and folinic acid (or D,L-leucovorin, or 5-formyl tetrahydrofolate), is a B-complex vitamin essential for normal CNS development and physiology. Abnormalities in folate levels (low CNS levels despite normal serum levels) and in folate-related pathways (genetic polymorphisms affecting enzymes involved in its metabolism) have been identified in children with autism, characterizing a condition known as cerebral folate deficiency syndrome (“CFD” – also found in schizophrenia and in other neurodevelopmental disorders) (https://pmc.ncbi.nlm.nih.gov/articles/PMC8622150/); https://www.degruyter.com/document/doi/10.1515/cclm-2012-0543/html).
The nervous system requires higher concentrations of folate than those found in the blood. To reach the nervous tissue, folates must cross the barriers that separate the blood from the CNS: the blood-brain barrier and the blood-cerebrospinal fluid barrier. To this end, the transport of folate to the CNS is essentially mediated by two highly specific systems (https://pubmed.ncbi.nlm.nih.gov/32200980/):
(1) A high-affinity transport mechanism (in which folate is transferred to nervous tissue at an energy expenditure) called the “folate receptor alpha”; this is the primary mechanism for folate transfer to the brain, capable of pumping folate to levels three times higher than those in the blood. Transport occurs through a mechanism called potocytosis, where the receptor-bound folate is internalized and then recycled back to the cell membrane.
(2) A transport mechanism that allows folates to be passively transferred to the CNS through the “reduced folate carrier” without using energy. This secondary system can only equalize brain folate levels with those in the blood. In other words, the concentrations of folate in the blood and the CNS are maintained in balance when this transport system is the sole option available.
What would be a third transporter mechanism (“proton-coupled”) actually functions as part of the mechanism mediated by the folate receptor alpha at the level of the blood-liquor barrier (https://pubmed.ncbi.nlm.nih.gov/19074442/).
Autistic children have high levels of autoantibodies directed against neurons, and the concentration of these antibodies directly correlates with the severity of their neuropsychiatric condition (https://pubmed.ncbi.nlm.nih.gov/22226851/). Several studies have shown that among autistic children, 70% have the antibody directed against the folate receptor (https://pubmed.ncbi.nlm.nih.gov/22230883/; https://pubmed.ncbi.nlm.nih.gov/20668945/).
Depending on the type of antibody produced, folate receptors are blocked or destroyed. The former block the folate-binding pocket, while binding antibodies can attach to other sites on the protein structure of the folate receptor alpha, followed by the attraction of the complement cascade, activation of cytokines, and finally destruction of the antigen-antibody complex (https://pubmed.ncbi.nlm.nih.gov/27068282/). The brain then becomes dependent on the second category of folate transporter (reduced folate transporter) to obtain a minimum level of folate, which is insufficient for the biological needs of neural cells. This condition (characterized by normal blood folate levels and reduced levels in cerebrospinal fluid – or “CSF”) has been termed “Cerebral Folate Deficiency” (CFD) syndrome and has also been found in other neurological and neuropsychiatric disorders, such as Rett syndrome, psychosis, refractory (intractable) schizophrenia, schizoaffective disorders, treatment-resistant major depression in adults, spastic-ataxic syndrome, and intractable epilepsy among young children (https://pubmed.ncbi.nlm.nih.gov/27068282/).
CFD results in impairment of the metabolic pathways used in the synthesis of nucleic acids (and therefore inhibition of neurogenesis—the formation of new neurons from stem cells residing in the CNS) (https://www.sciencedirect.com/science/article/pii/S2405844021021745). It also affects methylation processes, which are crucial for regulating gene expression, and hinders protective mechanisms against the damaging impacts of free radicals—cellular waste that must be continuously removed to prevent nerve cell damage (https://doi.org/10.1016/j.spen.2020.100835).
An important consequence of CFD is reduced glutathione synthesis. Glutathione is an important endogenous antioxidant that plays a crucial role in protecting cells against exogenous (such as heavy metals) and endogenous toxins, particularly in the CNS (https://pubmed.ncbi.nlm.nih.gov/22528835/). Abnormalities in reduced glutathione (GSH) metabolism result in oxidative damage to cellular DNA, proteins, and lipids. GSH abnormalities and markers of oxidative damage have been documented postmortem in brain regions involved in speech, emotion, and social behavior in individuals with autism (https://doi.org/10.1016/j.spen.2020.100835). In fact, methylation and redox abnormalities are so prevalent in autism that it has been proposed that their biomarkers be used for the diagnosis of ASD (https://doi.org/10.1016/j.spen.2020.100835).
Furthermore, the folate receptor alpha has functions independent of its role as a folate transporter; it also plays a role in maintaining the repertoire of stem cells from which new neurons must constantly originate (neurogenesis) both in prenatal life and throughout an individual’s lifespan (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5268765/). Its destruction by the antibodies found in high levels in autistic children, therefore, also compromises the recovery capacity of nervous tissue damaged by the inflammatory process that characterizes this condition.
Cerebral folate deficiency syndrome identified in autism can be treated with high doses of oral folinic acid (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7477301/). Folic acid supplementation is contraindicated and, if used, may aggravate CSF methylfolate deficiency (https://pubmed.ncbi.nlm.nih.gov/24494987/; https://pubmed.ncbi.nlm.nih.gov/20668945/). Elevated serum folate levels result in the transfer of this vitamin across the BBB via the “reduced folate transporter”. Elevated (supraphysiological) serum levels then provide restoration of the CNS’s own higher (physiological) concentrations.
In 2013, Frye et al. found that antibodies directed against the folate receptor alpha were present in 75% of children with autism when both blocking and binding antibodies were considered, with 29% being positive for both types of antibodies (https://pubmed.ncbi.nlm.nih.gov/22230883). Administration of folinic acid (leucovorin) at a dose of 2 mg per kg per day (maximum dose of 50 mg per day) provided improvements in communication, language, attention, and stereotypic behaviors in treated children compared to non-supplemented controls with ASD (https://pubmed.ncbi.nlm.nih.gov/22230883). This therapeutic approach was reviewed in 2020 (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7477301/), and the authors recommended gradually increasing the dose over the first 2 weeks to avoid a possible transient increase in agitation occasionally reported by parents. However, they themselves did not observe such a side effect; on the contrary, they found an improvement in excitement and agitation after approximately nine weeks of treatment. These results indicate that among autistic individuals who produce antibodies against the folate receptor alpha, some of the autistic manifestations may result from, or be aggravated by, cerebral folate deficiency syndrome, which can be corrected by administering high doses of folinic acid.
The presence of polymorphisms affecting the gene for an enzyme that plays a key role in folate metabolism (methylenetetrahydrofolate reductase, MTHFR) occurs in a significant percentage of the population (https://www.racgp.org.au/afp/2016/april/mthfr-genetic-testing-controversy-and-clinical-imp).
The presence of the C677T variant of the MTHFR gene, in particular, constitutes a risk factor for autism (https://journals.lww.com/psychgenetics/abstract/2009/08000/aberrations_in_folate_metabolic_pathway_and.2.aspx) and may interact with polymorphisms affecting other genes related to folate metabolism (such as the 19-base pair deletion polymorphism of the enzyme dihydrofolate reductase (DHFR) and the G80A SNP affecting the reduced folate carrier gene) to increase the risk of autism (https://pubmed.ncbi.nlm.nih.gov/17597297/). Evidently, these polymorphisms may exacerbate cerebral folate deficiency syndrome, particularly when they occur in association. The identification of a polymorphism affecting the DHFR gene as another risk factor for autism (https://pubmed.ncbi.nlm.nih.gov/17597297/) further emphasizes the need to avoid folic acid supplementation in autistic children, since the metabolism (reduction) of this synthetic folate requires DHFR activity.
MTHFR requires vitamin B2 (riboflavin), which acts as its cofactor (https://www.ncbi.nlm.nih.gov/books/NBK6145/). In turn, riboflavin deficiency may result from genetic factors prevalent in the general population (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1918332/), potentially representing an additional factor that contributes to susceptibility to autism. Thus, administering supraphysiological doses of riboflavin may prove to be beneficial.
In conclusion of this second Editorial, the evidence accumulated over the last decades makes clear the fundamental participation of autoimmune mechanisms in the pathophysiology of chronic encephalitis associated with autism (characterized in the first Editorial). The triad considered the “hallmark” of an autoimmune disease (inflammation, autoantibody production, and lymphocytic infiltration) is present in autism. In the next Editorial, we will see that the statement that “the origin of autoimmunity in autism is unknown” (https://pubmed.ncbi.nlm.nih.gov/22226851/ – page 465) is no longer supported.
Leia as postagens pelo índice mensal:
- outubro 2025
- julho 2025
- junho 2025
- abril 2025
- março 2025
- fevereiro 2025
- janeiro 2025
- dezembro 2024
- novembro 2024
- outubro 2024
- setembro 2024
- agosto 2024
- julho 2024
- julho 2021
- maio 2021
- abril 2021
- fevereiro 2021
- janeiro 2021
- dezembro 2020
- novembro 2020
- outubro 2020
- setembro 2020
- agosto 2020
- julho 2020
- junho 2020
- maio 2020
- abril 2020
Leia as últimas postagens:
- Evidencias científicas de que las vacunas actualmente usadas mundialmente causan Trastorno del Espectro Autista (TEA), un “Síndrome Autoinmune (Auto-inflamatorio) inducido por Adyuvantes” (ASIA). El mecanismo del “caballo de Troya”. Este es el tercer editorial de este sitio sobre la causa del autismo
 Primera editorial: https://wp.me/pbW3AH-1H7 Segundo editorial: https://wp.me/pbW3AH-1IX Traducido al español por Carmen Sanabria Ayala Índice 1) Introducción 2) Negacionismo 3) El impacto del autismo en el sistema educativo 4) Identificando los componentes de las vacunas como la causa raíz del TEA y el aumento del calendario de vacunación, incluidas las vacunaciones… Leia mais: Evidencias científicas de que las vacunas actualmente usadas mundialmente causan Trastorno del Espectro Autista (TEA), un “Síndrome Autoinmune (Auto-inflamatorio) inducido por Adyuvantes” (ASIA). El mecanismo del “caballo de Troya”. Este es el tercer editorial de este sitio sobre la causa del autismo
Primera editorial: https://wp.me/pbW3AH-1H7 Segundo editorial: https://wp.me/pbW3AH-1IX Traducido al español por Carmen Sanabria Ayala Índice 1) Introducción 2) Negacionismo 3) El impacto del autismo en el sistema educativo 4) Identificando los componentes de las vacunas como la causa raíz del TEA y el aumento del calendario de vacunación, incluidas las vacunaciones… Leia mais: Evidencias científicas de que las vacunas actualmente usadas mundialmente causan Trastorno del Espectro Autista (TEA), un “Síndrome Autoinmune (Auto-inflamatorio) inducido por Adyuvantes” (ASIA). El mecanismo del “caballo de Troya”. Este es el tercer editorial de este sitio sobre la causa del autismo - Norwegian version of the third editorial on the causes of autism
 Translation, Dr. Geir Flatabø Vitenskapelige bevis / evidens for at vaksiner i dag brukt over hele verden forårsaker Autisme Spekter Forstyrrelse (ASD) – en “Autoimmun (Auto Inflammatorisk) Syndrom Indusert av Adjuvans” (ASIA). “Trojansk hest” -mekanisme. “Stille infeksjon” i hjernevevet. Dette er den tredje redaksjonelle artikkel på dette nettstedet om autisme.… Leia mais: Norwegian version of the third editorial on the causes of autism
Translation, Dr. Geir Flatabø Vitenskapelige bevis / evidens for at vaksiner i dag brukt over hele verden forårsaker Autisme Spekter Forstyrrelse (ASD) – en “Autoimmun (Auto Inflammatorisk) Syndrom Indusert av Adjuvans” (ASIA). “Trojansk hest” -mekanisme. “Stille infeksjon” i hjernevevet. Dette er den tredje redaksjonelle artikkel på dette nettstedet om autisme.… Leia mais: Norwegian version of the third editorial on the causes of autism - Romanian version – Dovezi științifice că vaccinurile utilizate în prezent la nivel mondial cauzează tulburări din spectrul autist (TSA)
 Translated into romanian by Dr. Dominique Heidenfelder-Ambrosio Dovezi științifice că vaccinurile utilizate în prezent la nivel mondial cauzează tulburări din spectrul autist (TSA) – un „sindrom autoimun (autoinflamator) indus de adjuvanți” (ASIA). Mecanismul „calului troian”. „Infecția silențioasă” a țesutului cerebral. Acesta este al treilea editorial de pe acest site web… Leia mais: Romanian version – Dovezi științifice că vaccinurile utilizate în prezent la nivel mondial cauzează tulburări din spectrul autist (TSA)
Translated into romanian by Dr. Dominique Heidenfelder-Ambrosio Dovezi științifice că vaccinurile utilizate în prezent la nivel mondial cauzează tulburări din spectrul autist (TSA) – un „sindrom autoimun (autoinflamator) indus de adjuvanți” (ASIA). Mecanismul „calului troian”. „Infecția silențioasă” a țesutului cerebral. Acesta este al treilea editorial de pe acest site web… Leia mais: Romanian version – Dovezi științifice că vaccinurile utilizate în prezent la nivel mondial cauzează tulburări din spectrul autist (TSA) - Wissenschaftliche Erkenntnisse belegen, dass die derzeit weltweit verwendeten Impfstoffe Autismus Spektrum-Störungen verursachen
 Translated into German by Dr. Dominique Heidenfelder-Ambrosio Störungen (ASS) verursachen – ein durch Adjuvantien induziertes Autoimmunsyndrom (ASIA). Der „Trojanische Pferd“-Mechanismus. Eine „stille Infektion“ des Hirngewebes. Dies ist der dritte Leitartikel auf dieser Website zu den Ursachen von Autismus Index 1) Einleitung 2) Leugnung 3) Die Auswirkungen von Autismus auf das… Leia mais: Wissenschaftliche Erkenntnisse belegen, dass die derzeit weltweit verwendeten Impfstoffe Autismus Spektrum-Störungen verursachen
Translated into German by Dr. Dominique Heidenfelder-Ambrosio Störungen (ASS) verursachen – ein durch Adjuvantien induziertes Autoimmunsyndrom (ASIA). Der „Trojanische Pferd“-Mechanismus. Eine „stille Infektion“ des Hirngewebes. Dies ist der dritte Leitartikel auf dieser Website zu den Ursachen von Autismus Index 1) Einleitung 2) Leugnung 3) Die Auswirkungen von Autismus auf das… Leia mais: Wissenschaftliche Erkenntnisse belegen, dass die derzeit weltweit verwendeten Impfstoffe Autismus Spektrum-Störungen verursachen - Prove scientifiche che i vaccini attualmente utilizzati in tutto il mondo causano Disturbi dello Spettro Autistico
 Translated into Italian by Dr. Dominique Heidenfelder-Ambrosio “Sindrome Autoimmune (Auto-infiammatoria) indotta da Adiuvanti” (ASIA). Il meccanismo del “cavallo di Troia”. “Infezione silenziosa” del tessuto cerebrale. Questo è il terzo editoriale su questo sito web sulla causa dell’autismo Indice 1) Introduzione 2) Negazionismo 3) L’impatto dell’autismo sul sistema educativo 4) Identificare… Leia mais: Prove scientifiche che i vaccini attualmente utilizzati in tutto il mondo causano Disturbi dello Spettro Autistico
Translated into Italian by Dr. Dominique Heidenfelder-Ambrosio “Sindrome Autoimmune (Auto-infiammatoria) indotta da Adiuvanti” (ASIA). Il meccanismo del “cavallo di Troia”. “Infezione silenziosa” del tessuto cerebrale. Questo è il terzo editoriale su questo sito web sulla causa dell’autismo Indice 1) Introduzione 2) Negazionismo 3) L’impatto dell’autismo sul sistema educativo 4) Identificare… Leia mais: Prove scientifiche che i vaccini attualmente utilizzati in tutto il mondo causano Disturbi dello Spettro Autistico - Second editorial on the causes of autism, translated into norwegian
 Translated by Geir Flatabø En kronisk inflammatorisk prosess skader hjernen til barn som får en autisme diagnose, denne har en autoimmun komponent. En av konsekvensene er at transport av vitamin B9 (Folinsyre) til nervesystemet hemmes. Dette medfører alvorlig redusert funksjon i stoffskiftet som bruker vit B9, og selv om de… Leia mais: Second editorial on the causes of autism, translated into norwegian
Translated by Geir Flatabø En kronisk inflammatorisk prosess skader hjernen til barn som får en autisme diagnose, denne har en autoimmun komponent. En av konsekvensene er at transport av vitamin B9 (Folinsyre) til nervesystemet hemmes. Dette medfører alvorlig redusert funksjon i stoffskiftet som bruker vit B9, og selv om de… Leia mais: Second editorial on the causes of autism, translated into norwegian - Evidências científicas de que as vacinas atualmente usadas mundialmente causam Transtorno do Espectro Autista – Este é o terceiro editorial neste site sobre a causa do autismo
 Evidências científicas de que as vacinas atualmente usadas mundialmente causam Transtorno do Espectro Autista (TEA) – uma “Síndrome Autoimune (Auto-inflamatória) induzida por Adjuvantes” (ASIA). O mecanismo do “cavalo de Tróia”. Este é o terceiro editorial neste site sobre a causa do autismo. Índice 1) Introdução 2) Negacionismo 3) O impacto… Leia mais: Evidências científicas de que as vacinas atualmente usadas mundialmente causam Transtorno do Espectro Autista – Este é o terceiro editorial neste site sobre a causa do autismo
Evidências científicas de que as vacinas atualmente usadas mundialmente causam Transtorno do Espectro Autista (TEA) – uma “Síndrome Autoimune (Auto-inflamatória) induzida por Adjuvantes” (ASIA). O mecanismo do “cavalo de Tróia”. Este é o terceiro editorial neste site sobre a causa do autismo. Índice 1) Introdução 2) Negacionismo 3) O impacto… Leia mais: Evidências científicas de que as vacinas atualmente usadas mundialmente causam Transtorno do Espectro Autista – Este é o terceiro editorial neste site sobre a causa do autismo - Scientific evidence that vaccines currently used worldwide cause Autism Spectrum Disorder – Third editorial on this website on the cause of autism(Scientific evidence that vaccines currently used worldwide cause Autism Spectrum Disorder (ASD) – an “Autoimmune (Auto-inflammatory) Syndrome induced by Adjuvants” (ASIA). The “Trojan horse” mechanism. “Silent infection” of the brain tissue.) Index 1) Introduction 2) Denialism 3) The impact of autism on the education system 4) Identifying vaccine components as the… Leia mais: Scientific evidence that vaccines currently used worldwide cause Autism Spectrum Disorder – Third editorial on this website on the cause of autism
- Le processus inflammatoire chronique qui endommage le cerveau des enfants autistes présente une composante auto-immune incontestable – deuxième éditorial sur ce site concernant les causes de l’autisme
 Le processus inflammatoire chronique qui endommage le cerveau des enfants autistes présente une composante auto-immune incontestable. Parmi les conséquences, le transport de la vitamine B9 (folate) vers le système nerveux peut être inhibé, entraînant des implications thérapeutiques profondes qui, bien que documentées, ont été largement ignorées. Il s’agit du deuxième… Leia mais: Le processus inflammatoire chronique qui endommage le cerveau des enfants autistes présente une composante auto-immune incontestable – deuxième éditorial sur ce site concernant les causes de l’autisme
Le processus inflammatoire chronique qui endommage le cerveau des enfants autistes présente une composante auto-immune incontestable. Parmi les conséquences, le transport de la vitamine B9 (folate) vers le système nerveux peut être inhibé, entraînant des implications thérapeutiques profondes qui, bien que documentées, ont été largement ignorées. Il s’agit du deuxième… Leia mais: Le processus inflammatoire chronique qui endommage le cerveau des enfants autistes présente une composante auto-immune incontestable – deuxième éditorial sur ce site concernant les causes de l’autisme - Indicação de livro: Além da Vitamina D, versão em português
 Histórias emocionantes mostram como superar doenças autoimunes Mais do que um livro sobre saúde, este primeiro volume da série “Além da Vitamina D” relata histórias de pacientes, seus familiares, médicos, e médicos que são também pacientes do tratamento que devolve qualidade de vida a portadores de inúmeras doenças consideradas incuráveis.… Leia mais: Indicação de livro: Além da Vitamina D, versão em português
Histórias emocionantes mostram como superar doenças autoimunes Mais do que um livro sobre saúde, este primeiro volume da série “Além da Vitamina D” relata histórias de pacientes, seus familiares, médicos, e médicos que são também pacientes do tratamento que devolve qualidade de vida a portadores de inúmeras doenças consideradas incuráveis.… Leia mais: Indicação de livro: Além da Vitamina D, versão em português - Saiba as consequências da deficiência de Vitamina D – Dra. Satya Nahu
 Leia as postagens pelo índice mensal: Leia as últimas postagens:
Leia as postagens pelo índice mensal: Leia as últimas postagens: - Deficiência de vitamina D atende aos critérios de Hill para causalidade na suscetibilidade, complicações e mortalidade do SARS-CoV-2: uma revisão sistemática
 Fonte: https://www.mdpi.com/2072-6643/17/3/599 Os ensaios clínicos demonstram consistentemente uma correlação inversa entre os níveis séricos de 25-hidroxivitamina D [25(OH)D; calcifediol] e o risco de doença sintomática por SARS-CoV-2, complicações e mortalidade. Esta revisão sistemática (RS), guiada pelos critérios de causalidade de Bradford Hill, analisou 294 manuscritos revisados por pares publicados entre… Leia mais: Deficiência de vitamina D atende aos critérios de Hill para causalidade na suscetibilidade, complicações e mortalidade do SARS-CoV-2: uma revisão sistemática
Fonte: https://www.mdpi.com/2072-6643/17/3/599 Os ensaios clínicos demonstram consistentemente uma correlação inversa entre os níveis séricos de 25-hidroxivitamina D [25(OH)D; calcifediol] e o risco de doença sintomática por SARS-CoV-2, complicações e mortalidade. Esta revisão sistemática (RS), guiada pelos critérios de causalidade de Bradford Hill, analisou 294 manuscritos revisados por pares publicados entre… Leia mais: Deficiência de vitamina D atende aos critérios de Hill para causalidade na suscetibilidade, complicações e mortalidade do SARS-CoV-2: uma revisão sistemática - Norwegian version. Autisme hjernen er påvirket av en kronisk inflammatorisk prosess som kan og bør behandles. Dette er den første redaksjonelle artikkelen på denne nettsiden om årsakene til autismeTranslated by Geir Flatabø Autisme hjernen er påvirket av en kronisk inflammatorisk prosess som kan og bør behandles. Dette er den første redaksjonelle artikkelen på denne nettsiden om årsakene til autisme Autisme ble først beskrevet av Leo Kanner ( en barnepsykiater ved Johns Hopkins School of Medicine ) i 1943.… Leia mais: Norwegian version. Autisme hjernen er påvirket av en kronisk inflammatorisk prosess som kan og bør behandles. Dette er den første redaksjonelle artikkelen på denne nettsiden om årsakene til autisme
- “A Grande Indústria Farmacêutica é dona do seu médico”
 “A Big Pharma é dona do seu médico: “A coisa toda é bananas”. “Nossos médicos estudaram medicina em escolas financiadas pela Big Pharma. “A referência dos médicos científicos sobre a segurança e eficácia de vacinas e outros medicamentos é financiada pela Big Pharma. “As agências reguladoras de saúde que deveriam… Leia mais: “A Grande Indústria Farmacêutica é dona do seu médico”
“A Big Pharma é dona do seu médico: “A coisa toda é bananas”. “Nossos médicos estudaram medicina em escolas financiadas pela Big Pharma. “A referência dos médicos científicos sobre a segurança e eficácia de vacinas e outros medicamentos é financiada pela Big Pharma. “As agências reguladoras de saúde que deveriam… Leia mais: “A Grande Indústria Farmacêutica é dona do seu médico” - Como a vitamina D mudou a vida de uma médica com esclerose múltipla – sua jornada e uma entrevista -Protocolo CoimbraFonte: https://www.grassrootshealth.net/blog/vitamin-d-changed-life-doctor-ms-journey-interview/ “O Protocolo de Coimbra ajudou milhares de pessoas em todo o mundo a superar as suas doenças auto-imunes usando doses extremamente elevadas de vitamina D. Não se trate sem a orientação de um profissional qualificado.” “Narrativa de Satya L. Nahu, MD; não deixe de ler a curta entrevista… Leia mais: Como a vitamina D mudou a vida de uma médica com esclerose múltipla – sua jornada e uma entrevista -Protocolo Coimbra
- Dr. Toby Rogers: a aprovação do uso de alumínio em vacinas foi baseado em um estudo de apenas quatro coelhos
 “O FDA e o CDC aprovaram o alumínio como ‘seguro e eficaz’ em vacinas, com base em um estudo de apenas 4 coelhos.” Dr. Toby Rogers, PhD “Cada injeção no cronograma infantil do CDC, de acordo com o melhor conjunto de dados de vacinas do mundo, causa mais danos do… Leia mais: Dr. Toby Rogers: a aprovação do uso de alumínio em vacinas foi baseado em um estudo de apenas quatro coelhos
“O FDA e o CDC aprovaram o alumínio como ‘seguro e eficaz’ em vacinas, com base em um estudo de apenas 4 coelhos.” Dr. Toby Rogers, PhD “Cada injeção no cronograma infantil do CDC, de acordo com o melhor conjunto de dados de vacinas do mundo, causa mais danos do… Leia mais: Dr. Toby Rogers: a aprovação do uso de alumínio em vacinas foi baseado em um estudo de apenas quatro coelhos - Robert Kennedy Jr. acusa Congresso dos EUA de receber dinheiro da indústria farmacêutica em sua sabatina no Senado, no dia 29 de janeiro último – vídeo com legendas em português
 *Todas as publicações deste site são de exclusiva responsabilidade de seu criador e único administrador, Celso Galli Coimbra, OABRS 11352, e-mail cgcoimbra@gmail.com Leia as postagens pelo índice mensal: Links para as últimas 100 postagens: (Continua)
*Todas as publicações deste site são de exclusiva responsabilidade de seu criador e único administrador, Celso Galli Coimbra, OABRS 11352, e-mail cgcoimbra@gmail.com Leia as postagens pelo índice mensal: Links para as últimas 100 postagens: (Continua) - Estudo de “de cair o queixo” descobre que crianças vacinadas têm 170% maior risco de autismo
 Fonte:‘Jaw-dropping’ Study Finds Vaccinated Children Have 170% Higher Risk of Autism “O estudo revisado por pares também descobriu que as crianças vacinadas tinham uma probabilidade 212% maior de desenvolver outros distúrbios do neurodesenvolvimento, incluindo TDAH, epilepsia / apreensão, inflamação cerebral e distúrbios de tic e aprendizagem. As crianças vacinadas têm… Leia mais: Estudo de “de cair o queixo” descobre que crianças vacinadas têm 170% maior risco de autismo
Fonte:‘Jaw-dropping’ Study Finds Vaccinated Children Have 170% Higher Risk of Autism “O estudo revisado por pares também descobriu que as crianças vacinadas tinham uma probabilidade 212% maior de desenvolver outros distúrbios do neurodesenvolvimento, incluindo TDAH, epilepsia / apreensão, inflamação cerebral e distúrbios de tic e aprendizagem. As crianças vacinadas têm… Leia mais: Estudo de “de cair o queixo” descobre que crianças vacinadas têm 170% maior risco de autismo - Como trabalha o seu cérebro para a saúde ou para a doença – vídeo com legendas
 *Todas as publicações deste site são de exclusiva responsabilidade de seu criador e único administrador, Celso Galli Coimbra, OABRS 11352, e-mail cgcoimbra@gmail.com Leia as postagens pelo índice mensal: Links para as últimas 100 postagens:
*Todas as publicações deste site são de exclusiva responsabilidade de seu criador e único administrador, Celso Galli Coimbra, OABRS 11352, e-mail cgcoimbra@gmail.com Leia as postagens pelo índice mensal: Links para as últimas 100 postagens: - Anthony Fauci, um dos responsáveis pela “pandemia”, e pelo uso de vacinas como arma de guerra biológica, recebeu perdão antecipado nos últimos momentos do governo Biden – vídeo em português
 *Todas as publicações deste site são de exclusiva responsabilidade de seu criador e único administrador, Celso Galli Coimbra, OABRS 11352, e-mail cgcoimbra@gmail.com Leia as postagens pelo índice mensal: Links para as últimas 100 postagens:
*Todas as publicações deste site são de exclusiva responsabilidade de seu criador e único administrador, Celso Galli Coimbra, OABRS 11352, e-mail cgcoimbra@gmail.com Leia as postagens pelo índice mensal: Links para as últimas 100 postagens: - Vacinação e distúrbios do neurodesenvolvimento: um estudo de crianças de nove anos matriculadas no Medicaid – Transtorno do espectro autista. Publicação de 23.01.2025 na Science, Public Health Policy and the Law
 As taxas de distúrbios do neurodesenvolvimento (NDD) aumentaram mais de dez vezes durante a década de 1980 e uma em cada seis crianças dos EUA foi diagnosticada com uma deficiência de desenvolvimento entre 2009 e 2017. NDD é distúrbio do neurodesenvolvimento Leia na fonte da publicação: Vaccination and Neurodevelopmental Disorders:… Leia mais: Vacinação e distúrbios do neurodesenvolvimento: um estudo de crianças de nove anos matriculadas no Medicaid – Transtorno do espectro autista. Publicação de 23.01.2025 na Science, Public Health Policy and the Law
As taxas de distúrbios do neurodesenvolvimento (NDD) aumentaram mais de dez vezes durante a década de 1980 e uma em cada seis crianças dos EUA foi diagnosticada com uma deficiência de desenvolvimento entre 2009 e 2017. NDD é distúrbio do neurodesenvolvimento Leia na fonte da publicação: Vaccination and Neurodevelopmental Disorders:… Leia mais: Vacinação e distúrbios do neurodesenvolvimento: um estudo de crianças de nove anos matriculadas no Medicaid – Transtorno do espectro autista. Publicação de 23.01.2025 na Science, Public Health Policy and the Law - Múltiplas vacinações simultâneas estão associadas à probabilidade de hospitalização ou morte por uma reação adversa
 (Todas as publicações deste site são de exclusiva responsabilidade de seu criador e único administrador, Celso Galli Coimbra, OABRS 11352, e-mail cgcoimbra@gmail.com) Múltiplas vacinações simultâneas (comumente quatro a oito injeções) são aplicadas de forma recorrente a intervalos tão curtos quanto dois meses ao longo dos primeiros anos de vida (https://www.cdc.gov/vaccines/hcp/imz-schedules/child-adolescent-age.html). Nenhuma… Leia mais: Múltiplas vacinações simultâneas estão associadas à probabilidade de hospitalização ou morte por uma reação adversa
(Todas as publicações deste site são de exclusiva responsabilidade de seu criador e único administrador, Celso Galli Coimbra, OABRS 11352, e-mail cgcoimbra@gmail.com) Múltiplas vacinações simultâneas (comumente quatro a oito injeções) são aplicadas de forma recorrente a intervalos tão curtos quanto dois meses ao longo dos primeiros anos de vida (https://www.cdc.gov/vaccines/hcp/imz-schedules/child-adolescent-age.html). Nenhuma… Leia mais: Múltiplas vacinações simultâneas estão associadas à probabilidade de hospitalização ou morte por uma reação adversa - Reconsideração dos níveis de dose segura pediátrica imunoterapêutica de alumínio – Reconsideration of the immunotherapeutic pediatric safe dose levels of aluminum
 Fonte: https://www.sciencedirect.com/science/article/pii/S0946672X17300950#bib0210 Conclusões do artigo: “Os níveis de alumínio nas vacinas são baseados na eficácia imunológica e ignoram o cálculo da segurança conforme o peso corporal.” “Vários erros críticos foram cometidos na avaliação da dosagem pediátrica de alumínio em vacinas.” “As inferências de segurança das doses de de alumínio em… Leia mais: Reconsideração dos níveis de dose segura pediátrica imunoterapêutica de alumínio – Reconsideration of the immunotherapeutic pediatric safe dose levels of aluminum
Fonte: https://www.sciencedirect.com/science/article/pii/S0946672X17300950#bib0210 Conclusões do artigo: “Os níveis de alumínio nas vacinas são baseados na eficácia imunológica e ignoram o cálculo da segurança conforme o peso corporal.” “Vários erros críticos foram cometidos na avaliação da dosagem pediátrica de alumínio em vacinas.” “As inferências de segurança das doses de de alumínio em… Leia mais: Reconsideração dos níveis de dose segura pediátrica imunoterapêutica de alumínio – Reconsideration of the immunotherapeutic pediatric safe dose levels of aluminum - Alumínio no tecido cerebral no autismo – Aluminium in brain tissue in autism – ScienceDirect
 Fonte: https://www.sciencedirect.com/science/article/pii/S0946672X17308763 “…o fato de termos encontrado alumínio em cada amostra de tecido cerebral, congelada ou fixada, sugere fortemente que os indivíduos com diagnóstico de Transtorno do Espectro Autista têm níveis extraordinariamente altos de alumínio em seu tecido cerebral e que este alumínio está particularmente associado a células não neuronais… Leia mais: Alumínio no tecido cerebral no autismo – Aluminium in brain tissue in autism – ScienceDirect
Fonte: https://www.sciencedirect.com/science/article/pii/S0946672X17308763 “…o fato de termos encontrado alumínio em cada amostra de tecido cerebral, congelada ou fixada, sugere fortemente que os indivíduos com diagnóstico de Transtorno do Espectro Autista têm níveis extraordinariamente altos de alumínio em seu tecido cerebral e que este alumínio está particularmente associado a células não neuronais… Leia mais: Alumínio no tecido cerebral no autismo – Aluminium in brain tissue in autism – ScienceDirect - Autystyczny mózg jest uszkodzony przez nieleczony przewlekły proces zapalny. Pierwsza publikacja na tej stronie o przyczynach autyzmu
 Translated into Polish by Father Jacek Maria Norkowski OP, Md. PhD., former teacher of moral theology in Pontifical University of Saint Thomas Aquinas in Rome. Autyzm został po raz pierwszy opisany przez Leo Kannera (Psychiatra dziecięcy w Johns Hopkins School of Medicine) w 1943 roku, który nazwał to “zaburzeniami autystycznymi”(https://www.autismtruths.org/pdf/autistic%20disturbances%20of%20affective%20contact%20-%20leo%20kanner.pdf).… Leia mais: Autystyczny mózg jest uszkodzony przez nieleczony przewlekły proces zapalny. Pierwsza publikacja na tej stronie o przyczynach autyzmu
Translated into Polish by Father Jacek Maria Norkowski OP, Md. PhD., former teacher of moral theology in Pontifical University of Saint Thomas Aquinas in Rome. Autyzm został po raz pierwszy opisany przez Leo Kannera (Psychiatra dziecięcy w Johns Hopkins School of Medicine) w 1943 roku, który nazwał to “zaburzeniami autystycznymi”(https://www.autismtruths.org/pdf/autistic%20disturbances%20of%20affective%20contact%20-%20leo%20kanner.pdf).… Leia mais: Autystyczny mózg jest uszkodzony przez nieleczony przewlekły proces zapalny. Pierwsza publikacja na tej stronie o przyczynach autyzmu - Der chronische Entzündungsprozess, der das Gehirn autistischer Kinder betrifft, hat unbestreitbar eine Autoimmunkomponente – Zweiter Leitartikel auf dieser Website zu den Ursachen von AutismusBewertet von:Dr. Melanie WeissWilhelmstraße 17342489 Wülfrathinfo@neurologie-wuelfrath.de (Erster ins Deutsche übersetzter Leitartikel: https://wp.me/pbW3AH-1Gu) Der chronische Entzündungsprozess, der das Gehirn autistischer Kinder schädigt, weist eindeutig eine autoimmune Komponente auf. Infolgedessen wird der Transport von Vitamin B9 (Folat) zum Nervensystem gehemmt, was zu tiefgreifenden Auswirkungen führen kann, die zwar dokumentiert, aber weitgehend ignoriert… Leia mais: Der chronische Entzündungsprozess, der das Gehirn autistischer Kinder betrifft, hat unbestreitbar eine Autoimmunkomponente – Zweiter Leitartikel auf dieser Website zu den Ursachen von Autismus
- O papel da vitamina D nos resultados dos cuidados intensivos em pacientes com COVID-19
 Fonte: O papel da vitamina D nos resultados dos cuidados intensivos em pacientes com COVID-19: evidência de uma meta-análise guarda-chuva de estudos intervencionistas e observacionais Publicado online pela Cambridge University Press: 24 de Abril de 2024 Conclusão do estudo “No geral, o estado da vitamina D é um fator crítico… Leia mais: O papel da vitamina D nos resultados dos cuidados intensivos em pacientes com COVID-19
Fonte: O papel da vitamina D nos resultados dos cuidados intensivos em pacientes com COVID-19: evidência de uma meta-análise guarda-chuva de estudos intervencionistas e observacionais Publicado online pela Cambridge University Press: 24 de Abril de 2024 Conclusão do estudo “No geral, o estado da vitamina D é um fator crítico… Leia mais: O papel da vitamina D nos resultados dos cuidados intensivos em pacientes com COVID-19 - Robert Kennedy Jr denunciou que a indústria farmacêutica sabia que suas vacinas causavam autismo – vídeo com legendas
 De acordo com RFK Jr., uma denunciante lhe forneceu uma transcrição de uma reunião secreta envolvendo autoridades do FDA, CDC, NIH e OMS, provando que as autoridades de saúde pública sabiam que as vacinas infantis estavam causando autismo. “Eles observaram a vacina contra hepatite B administrada durante os primeiros 30… Leia mais: Robert Kennedy Jr denunciou que a indústria farmacêutica sabia que suas vacinas causavam autismo – vídeo com legendas
De acordo com RFK Jr., uma denunciante lhe forneceu uma transcrição de uma reunião secreta envolvendo autoridades do FDA, CDC, NIH e OMS, provando que as autoridades de saúde pública sabiam que as vacinas infantis estavam causando autismo. “Eles observaram a vacina contra hepatite B administrada durante os primeiros 30… Leia mais: Robert Kennedy Jr denunciou que a indústria farmacêutica sabia que suas vacinas causavam autismo – vídeo com legendas - Os adjuvantes de vacinas de alumínio contribuem para o aumento da prevalência do autismo?
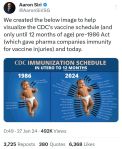 “os recém-nascidos recebem uma dose de alumínio equivalente a 10 (dez) vacinas contra hepatite B administradas simultaneamente a um adulto de 70 kg” Tabela número 5, extraída da publicação “Do aluminum vaccine adjuvants contribute to the rising prevalence of autism? (Os adjuvantes* de vacinas de alumínio contribuem para o aumento… Leia mais: Os adjuvantes de vacinas de alumínio contribuem para o aumento da prevalência do autismo?
“os recém-nascidos recebem uma dose de alumínio equivalente a 10 (dez) vacinas contra hepatite B administradas simultaneamente a um adulto de 70 kg” Tabela número 5, extraída da publicação “Do aluminum vaccine adjuvants contribute to the rising prevalence of autism? (Os adjuvantes* de vacinas de alumínio contribuem para o aumento… Leia mais: Os adjuvantes de vacinas de alumínio contribuem para o aumento da prevalência do autismo? - Il processo infiammatorio cronico che sta vivendo il cervello dei bambini autistici ha un’innegabile componente autoimmune – Secondo Editoriale di questo Sito sulle cause dell’autismoTraduzione fatta da Dottoressa Francesca Micheluccifmichelucci22@gmail.com, Pisa Primo editoriale tradotto in italiano https://wp.me/pbW3AH-1FM Il processo infiammatorio cronico che danneggia il cervello dei bambini autistici presenta un’innegabile componente autoimmune. Tra le varie conseguenze, può essere inibito il trasporto della vitamina B9 ( folato ) attraverso il sistema nervoso , portando a… Leia mais: Il processo infiammatorio cronico che sta vivendo il cervello dei bambini autistici ha un’innegabile componente autoimmune – Secondo Editoriale di questo Sito sulle cause dell’autismo
- As chantagens da Pfizer para impor sua vacina aos países durante a pandemia – Pfizer’s blackmail to impose its vaccine on countries during the pandemic – com legendas
 Leia as postagens pelo índice mensal: Leia as últimas postagens:
Leia as postagens pelo índice mensal: Leia as últimas postagens: - Segundo Editorial sobre las Causas de Autismo, traducido al español. El proceso inflamatorio crónico que está dañando el cerebro de los niños autistas tiene un innegable componente autoinmune.(Primera editorial: https://wp.me/pbW3AH-1H7) Traducido al español por Carmen Sanabria Ayala Entre las consecuencias está la inhibición del transporte de vitamina B9 (folato) al sistema nervioso, con profundas implicaciones terapéuticas que, a pesar de estar documentadas, han sido en gran medida ignoradas. Segundo Editorial de este Site sobre las causas del… Leia mais: Segundo Editorial sobre las Causas de Autismo, traducido al español. El proceso inflamatorio crónico que está dañando el cerebro de los niños autistas tiene un innegable componente autoinmune.
- Uma associação positiva encontrada entre a prevalência do autismo e a aceitação da vacinação infantil em toda a população dos EUA
 Fonte: https://pubmed.ncbi.nlm.nih.gov/21623535/ NIH, National Library of Medicine, “Um site oficial do governo dos Estados Unidos” Publicação de 2011 PMID: Endereço de 21623535DOI: 10.1080/15287394.2011.573736 “Abstrato “A razão para o rápido aumento do autismo nos Estados Unidos que começou na década de 1990 é um mistério. Embora os indivíduos provavelmente tenham uma… Leia mais: Uma associação positiva encontrada entre a prevalência do autismo e a aceitação da vacinação infantil em toda a população dos EUA
Fonte: https://pubmed.ncbi.nlm.nih.gov/21623535/ NIH, National Library of Medicine, “Um site oficial do governo dos Estados Unidos” Publicação de 2011 PMID: Endereço de 21623535DOI: 10.1080/15287394.2011.573736 “Abstrato “A razão para o rápido aumento do autismo nos Estados Unidos que começou na década de 1990 é um mistério. Embora os indivíduos provavelmente tenham uma… Leia mais: Uma associação positiva encontrada entre a prevalência do autismo e a aceitação da vacinação infantil em toda a população dos EUA - Estudos publicados que mostram que vacinas causam autismo – Studies published showing vaccines cause autism
 Studies which show vaccines cause Autism: ▪ http://ncbi.nlm.nih.gov/pmc/articles/PMC3878266/ ▪ http://ncbi.nlm.nih.gov/pubmed/21623535 ▪ http://ncbi.nlm.nih.gov/pubmed/25377033 ▪ http://ncbi.nlm.nih.gov/pubmed/24995277 ▪ http://ncbi.nlm.nih.gov/pubmed/12145534 ▪ http://ncbi.nlm.nih.gov/pubmed/21058170 ▪ http://ncbi.nlm.nih.gov/pubmed/22099159 ▪ http://ncbi.nlm.nih.gov/pmc/articles/PMC3364648/ ▪ http://ncbi.nlm.nih.gov/pubmed/17454560 ▪ http://ncbi.nlm.nih.gov/pubmed/19106436 ▪ http://ncbi.nlm.nih.gov/pmc/articles/PMC3774468/ ▪ http://ncbi.nlm.nih.gov/pmc/articles/PMC3697751/ ▪ http://ncbi.nlm.nih.gov/pubmed/21299355 ▪ http://ncbi.nlm.nih.gov/pubmed/21907498 ▪ http://ncbi.nlm.nih.gov/pubmed/11339848 ▪ http://ncbi.nlm.nih.gov/pubmed/17674242 ▪ http://ncbi.nlm.nih.gov/pubmed/21993250 ▪ http://ncbi.nlm.nih.gov/pubmed/15780490 ▪ http://ncbi.nlm.nih.gov/pubmed/12933322 ▪ http://ncbi.nlm.nih.gov/pubmed/16870260 ▪ http://ncbi.nlm.nih.gov/pubmed/19043938 ▪ http://ncbi.nlm.nih.gov/pubmed/12142947… Leia mais: Estudos publicados que mostram que vacinas causam autismo – Studies published showing vaccines cause autism
Studies which show vaccines cause Autism: ▪ http://ncbi.nlm.nih.gov/pmc/articles/PMC3878266/ ▪ http://ncbi.nlm.nih.gov/pubmed/21623535 ▪ http://ncbi.nlm.nih.gov/pubmed/25377033 ▪ http://ncbi.nlm.nih.gov/pubmed/24995277 ▪ http://ncbi.nlm.nih.gov/pubmed/12145534 ▪ http://ncbi.nlm.nih.gov/pubmed/21058170 ▪ http://ncbi.nlm.nih.gov/pubmed/22099159 ▪ http://ncbi.nlm.nih.gov/pmc/articles/PMC3364648/ ▪ http://ncbi.nlm.nih.gov/pubmed/17454560 ▪ http://ncbi.nlm.nih.gov/pubmed/19106436 ▪ http://ncbi.nlm.nih.gov/pmc/articles/PMC3774468/ ▪ http://ncbi.nlm.nih.gov/pmc/articles/PMC3697751/ ▪ http://ncbi.nlm.nih.gov/pubmed/21299355 ▪ http://ncbi.nlm.nih.gov/pubmed/21907498 ▪ http://ncbi.nlm.nih.gov/pubmed/11339848 ▪ http://ncbi.nlm.nih.gov/pubmed/17674242 ▪ http://ncbi.nlm.nih.gov/pubmed/21993250 ▪ http://ncbi.nlm.nih.gov/pubmed/15780490 ▪ http://ncbi.nlm.nih.gov/pubmed/12933322 ▪ http://ncbi.nlm.nih.gov/pubmed/16870260 ▪ http://ncbi.nlm.nih.gov/pubmed/19043938 ▪ http://ncbi.nlm.nih.gov/pubmed/12142947… Leia mais: Estudos publicados que mostram que vacinas causam autismo – Studies published showing vaccines cause autism - First editorial, translated into Russian. Хронический воспалительный процесс в мозге, – причина Расстройств Аутистического Спектра, требующая обязательного лечения
 Russian translator Dr Ph.D. Sementsov Vadim AlexandrovichInsta: @doctorsementsov Первое редакционное сообщение о причинах аутизма. Профессор Цицеро Гали Коимбра 2024 г. Аутизм был впервые описан Лео Каннером (детским психиатром из Медицинской школы Джона Хопкинса) в 1943 году, который назвал его «аутистическими расстройствами». Девять лет спустя стала очевидной степень тяжести этого поведенческого… Leia mais: First editorial, translated into Russian. Хронический воспалительный процесс в мозге, – причина Расстройств Аутистического Спектра, требующая обязательного лечения
Russian translator Dr Ph.D. Sementsov Vadim AlexandrovichInsta: @doctorsementsov Первое редакционное сообщение о причинах аутизма. Профессор Цицеро Гали Коимбра 2024 г. Аутизм был впервые описан Лео Каннером (детским психиатром из Медицинской школы Джона Хопкинса) в 1943 году, который назвал его «аутистическими расстройствами». Девять лет спустя стала очевидной степень тяжести этого поведенческого… Leia mais: First editorial, translated into Russian. Хронический воспалительный процесс в мозге, – причина Расстройств Аутистического Спектра, требующая обязательного лечения - Pagamentos da indústria farmacêutica e dispositivos médicos para revisores de pares dos EUA de grandes revistas médicas supera um bilhão de dólares
 Entre 2020 e 2022, a indústria farmacêutica destinou cerca de US$ 1,06 bilhão a revisores de grandes revistas médicas, incluindo The BMJ, JAMA, The Lancet e The New England Journal of Medicine. Essa informação já esteve postada por outra fonte do meio científico neste site, em 2020. Continua sendo denunciada… Leia mais: Pagamentos da indústria farmacêutica e dispositivos médicos para revisores de pares dos EUA de grandes revistas médicas supera um bilhão de dólares
Entre 2020 e 2022, a indústria farmacêutica destinou cerca de US$ 1,06 bilhão a revisores de grandes revistas médicas, incluindo The BMJ, JAMA, The Lancet e The New England Journal of Medicine. Essa informação já esteve postada por outra fonte do meio científico neste site, em 2020. Continua sendo denunciada… Leia mais: Pagamentos da indústria farmacêutica e dispositivos médicos para revisores de pares dos EUA de grandes revistas médicas supera um bilhão de dólares - ऑटिस्टिक मस्तिष्क एक अनुपचारित दीर्घकालिक सूजन प्रक्रिया से प्रभावित रहता है। इस वेबसाइट पर ऑटिज़्म के कारणों के बारे में यह पहला संपादकीय लेख है। – First editorial, hindi version: The autistic brain is affected by an untreated chronic inflammatory process
 Translated from English into Hindi by Dr. Renu Mahtani(अंग्रेजी से हिंदी में डॉ. रेणु महतानी द्वारा अनुवादित)Param Health Centre, Pune, Indiaparamhealth@gmail.com “ऑटिस्टिक मस्तिष्क एक अनुपचारित दीर्घकालिक सूजन प्रक्रिया से प्रभावित रहता है। इस वेबसाइट पर ऑटिज़्म के कारणों के बारे में यह पहला संपादकीय लेख है।” ऑटिज़्म का पहली बार… Leia mais: ऑटिस्टिक मस्तिष्क एक अनुपचारित दीर्घकालिक सूजन प्रक्रिया से प्रभावित रहता है। इस वेबसाइट पर ऑटिज़्म के कारणों के बारे में यह पहला संपादकीय लेख है। – First editorial, hindi version: The autistic brain is affected by an untreated chronic inflammatory process
Translated from English into Hindi by Dr. Renu Mahtani(अंग्रेजी से हिंदी में डॉ. रेणु महतानी द्वारा अनुवादित)Param Health Centre, Pune, Indiaparamhealth@gmail.com “ऑटिस्टिक मस्तिष्क एक अनुपचारित दीर्घकालिक सूजन प्रक्रिया से प्रभावित रहता है। इस वेबसाइट पर ऑटिज़्म के कारणों के बारे में यह पहला संपादकीय लेख है।” ऑटिज़्म का पहली बार… Leia mais: ऑटिस्टिक मस्तिष्क एक अनुपचारित दीर्घकालिक सूजन प्रक्रिया से प्रभावित रहता है। इस वेबसाइट पर ऑटिज़्म के कारणों के बारे में यह पहला संपादकीय लेख है। – First editorial, hindi version: The autistic brain is affected by an untreated chronic inflammatory process - Nos últimos anos, as indústrias de vacinas pagaram 35 bilhões de dólares em multas por falsificação de informações aos médicos, mas estão protegidas das consequências criminais
 *Todas as publicações deste site são de exclusiva responsabilidade de seu criador e único administrador, Celso Galli Coimbra, OABRS 11352, e-mail cgcoimbra@gmail.com Leia as postagens pelo índice mensal: Leia as últimas postagens:
*Todas as publicações deste site são de exclusiva responsabilidade de seu criador e único administrador, Celso Galli Coimbra, OABRS 11352, e-mail cgcoimbra@gmail.com Leia as postagens pelo índice mensal: Leia as últimas postagens: - Análise de Robert Kennedy Jr. sobre a saúde nos Estados Unidos, especialmente de crianças e jovens, e as Agências Reguladoras – com legendas
 Leia as postagens pelo índice mensal: Leia as últimas postagens:
Leia as postagens pelo índice mensal: Leia as últimas postagens: - El cerebro autista se ve afectado por un proceso inflamatorio crónico no tratado. Primer Editorial en este Sitio sobre las causas del autismo, actualizado el 06/10/2024Traducido al español por Carmen Sanabria Ayala El autismo fue descrito por primera vez por Leo Kanner (psiquiatra infantil de la Escuela de Medicina Johns Hopkins) en 1943, quien lo llamó de “trastornos autistas” (https://www.autismtruths.org/pdf/Autistic%20Disturbances%20of%20Affective%20Contact%20-%20Leo%20Kanner.pdf). Nueve años más tarde, la gravedad que esta condición conductual podría alcanzar se hizo evidente… Leia mais: El cerebro autista se ve afectado por un proceso inflamatorio crónico no tratado. Primer Editorial en este Sitio sobre las causas del autismo, actualizado el 06/10/2024
- Trabalhadores de trânsito dos EUA demitidos por recusarem vacina contra a COVID-19 receberão mais de US$ 1 milhão cada
 *Todas as publicações deste site são escolha de exclusiva responsabilidade de seu criador e único administrador, Celso Galli Coimbra, OABRS 11352, e-mail cgcoimbra@gmail.com Leia as postagens pelo índice mensal: Leia as últimas postagens:
*Todas as publicações deste site são escolha de exclusiva responsabilidade de seu criador e único administrador, Celso Galli Coimbra, OABRS 11352, e-mail cgcoimbra@gmail.com Leia as postagens pelo índice mensal: Leia as últimas postagens: - Romanian version: Procesul inflamator cronic, care dăunează creierului copiilor cu autism are o componentă autoimună incontestabilă. Al doilea Editorial de pe acest site despre cauzele autismuluiRomanian translation by Dr. Dominique Heidenfelder-Ambrosio Bazându-se pe logica luminată de publicațiile științifice, care s-au acumulat de-a lungul deceniilor, această serie de Editoriale își propune să ofere cititorului o cale de identificare a etiologiei (cauzei) autismului — o afecțiune al cărei cost anual, inclusiv cheltuielile directe și indirecte, ar putea… Leia mais: Romanian version: Procesul inflamator cronic, care dăunează creierului copiilor cu autism are o componentă autoimună incontestabilă. Al doilea Editorial de pe acest site despre cauzele autismului
- Autistični mozak zahvaćen je neliječenim kroničnim upalnim procesom. Prvi post na ovoj web stranici govori o uzrocima autizma
 Prijevod s engleskog na hrvatski Goranka Tomić i dr. Fedor Mataić Autizam je prvi opisao Leo Kanner (dječji psihijatar na Medicinskom fakultetu The Johns Hopkins) 1943. godine, koji ga je nazvao “autistični poremećaji”. (https://www.autismtruths.org/pdf/Autistic%20Disturbances%20of%20Affective%20Contact%20-%20Leo%20Kanner.pdf). Devet godina kasnije, ozbiljnost koju bi to stanje ponašanja moglo doseći postala je očigledna kada je… Leia mais: Autistični mozak zahvaćen je neliječenim kroničnim upalnim procesom. Prvi post na ovoj web stranici govori o uzrocima autizma
Prijevod s engleskog na hrvatski Goranka Tomić i dr. Fedor Mataić Autizam je prvi opisao Leo Kanner (dječji psihijatar na Medicinskom fakultetu The Johns Hopkins) 1943. godine, koji ga je nazvao “autistični poremećaji”. (https://www.autismtruths.org/pdf/Autistic%20Disturbances%20of%20Affective%20Contact%20-%20Leo%20Kanner.pdf). Devet godina kasnije, ozbiljnost koju bi to stanje ponašanja moglo doseći postala je očigledna kada je… Leia mais: Autistični mozak zahvaćen je neliječenim kroničnim upalnim procesom. Prvi post na ovoj web stranici govori o uzrocima autizma - Autismus – ein unbehandelter chronischer Entzündungsprozess des Gehirns. Erster Leitartikel auf dieser Website über die Ursachen von AutismusÜbersetzung aus der englischen Version: Prof. Dr. Nelson AnnunciatoNeurowissenschaftler Bewertet von:Dr. Melanie WeissWilhelmstraße 17342489 Wülfrathinfo@neurologie-wuelfrath.de Autismus wurde erstmals 1943 von Leo Kanner (einem Kinderpsychiater an der Johns Hopkins School of Medicine) beschrieben, der es als „autistische Störung “bezeichnete (https://www.autismtruths.org/pdf/Autistic%20Disturbances%20of%20Affective%20Contact%20-%20Leo%20Kanner.pdf). Neun Jahre später wurde die Schwere, die dieser Verhaltenszustand erreichen kann,… Leia mais: Autismus – ein unbehandelter chronischer Entzündungsprozess des Gehirns. Erster Leitartikel auf dieser Website über die Ursachen von Autismus
- The chronic inflammatory process that is damaging the brains of autistic children has an undeniable autoimmune component. The second editorial on this site on the causes of autism
 (Link to the first editorial: https://wp.me/pbW3AH-1DO -Link to the third editorial of this series: https://wp.me/pbW3AH-1Tx) Based on the logic enlightened by scientific publications that have accumulated over decades, this series of editorials aims to offer the reader a path to identifying the etiology (cause) of autism—a condition whose annual cost,… Leia mais: The chronic inflammatory process that is damaging the brains of autistic children has an undeniable autoimmune component. The second editorial on this site on the causes of autism
(Link to the first editorial: https://wp.me/pbW3AH-1DO -Link to the third editorial of this series: https://wp.me/pbW3AH-1Tx) Based on the logic enlightened by scientific publications that have accumulated over decades, this series of editorials aims to offer the reader a path to identifying the etiology (cause) of autism—a condition whose annual cost,… Leia mais: The chronic inflammatory process that is damaging the brains of autistic children has an undeniable autoimmune component. The second editorial on this site on the causes of autism - French version. Le cerveau des personnes autistes est affecté par un processus inflammatoire chronique non traité. Premier éditorial sur ce site web concernant les causes de l’autisme
 Traduction en français par Dr. Djamel Deramchi L’autisme a été décrit pour la première fois par Leo Kanner (pédopsychiatre à la faculté de médecine Johns Hopkins) en 1943, qui l’a qualifié de « troubles autistiques » (https://www.autismtruths.org/pdf/Autistic%20Disturbances%20of%20Affective%20Contact%20-%20Leo%20Kanner.pdf). Neuf ans plus tard, la gravité que cette condition comportementale pouvait atteindre est devenue évidente… Leia mais: French version. Le cerveau des personnes autistes est affecté par un processus inflammatoire chronique non traité. Premier éditorial sur ce site web concernant les causes de l’autisme
Traduction en français par Dr. Djamel Deramchi L’autisme a été décrit pour la première fois par Leo Kanner (pédopsychiatre à la faculté de médecine Johns Hopkins) en 1943, qui l’a qualifié de « troubles autistiques » (https://www.autismtruths.org/pdf/Autistic%20Disturbances%20of%20Affective%20Contact%20-%20Leo%20Kanner.pdf). Neuf ans plus tard, la gravité que cette condition comportementale pouvait atteindre est devenue évidente… Leia mais: French version. Le cerveau des personnes autistes est affecté par un processus inflammatoire chronique non traité. Premier éditorial sur ce site web concernant les causes de l’autisme - Il cervello del paziente autistico è affetto da un processo infiammatorio cronico non trattato. Su questo sito web primo Editoriale sulle cause dell’autismoTtradotto da Dottssa Giusi Manuele Specialista Endocrinochirurgia Protocollo Coimbra Italia L’autismo fu descritto per la prima volta da Leo Kanner (psichiatra infantile della Johns Hopkins School of Medicine) nel 1943, che lo chiamò “disturbo autistico” (https://www.autismtruths.org/pdf/Autistic%20Disturbances%20of%20Affective%20Contact%20-%20Leo%20Kanner.pdf). Nove anni dopo, si comprese la gravità che questa condizione comportamentale poteva raggiungere e… Leia mais: Il cervello del paziente autistico è affetto da un processo infiammatorio cronico non trattato. Su questo sito web primo Editoriale sulle cause dell’autismo
- Enquanto o Reino Unido debate a legalização do suicídio assistido, a prática aumenta drasticamente no Canadá
 Depois que as eleições de 4 de julho no Reino Unido fizeram de Keir Starmer o novo primeiro-ministro, o suicídio assistido virou notícia. A reportagem é de Charles Collins, publicada por Crux, 13-08-2024. No início deste ano, Starmer disse que estava pessoalmente comprometido em mudar as leis do país que tornam o suicídio… Leia mais: Enquanto o Reino Unido debate a legalização do suicídio assistido, a prática aumenta drasticamente no Canadá
Depois que as eleições de 4 de julho no Reino Unido fizeram de Keir Starmer o novo primeiro-ministro, o suicídio assistido virou notícia. A reportagem é de Charles Collins, publicada por Crux, 13-08-2024. No início deste ano, Starmer disse que estava pessoalmente comprometido em mudar as leis do país que tornam o suicídio… Leia mais: Enquanto o Reino Unido debate a legalização do suicídio assistido, a prática aumenta drasticamente no Canadá - Geração Z e Mortes do Desespero
 O professor de SPA analisa as tendências alarmantes do suicídio nacional, incluindo um aumento sem precedentes entre os jovens, e analisa possíveis determinantes. 10 de Agosto, 2023 Depois de cair por décadas, a taxa de suicídio nos Estados Unidos aumentou quase 40% desde 2000. Em “The Re-Emerging Suicide Crisis in… Leia mais: Geração Z e Mortes do Desespero
O professor de SPA analisa as tendências alarmantes do suicídio nacional, incluindo um aumento sem precedentes entre os jovens, e analisa possíveis determinantes. 10 de Agosto, 2023 Depois de cair por décadas, a taxa de suicídio nos Estados Unidos aumentou quase 40% desde 2000. Em “The Re-Emerging Suicide Crisis in… Leia mais: Geração Z e Mortes do Desespero - Estudo mostra taxas de suicídio e autolesão entre jovens em alta no Brasil
 Fonte: https://portal.fiocruz.br/en/news/2024/02/study-shows-suicide-and-self-injury-rates-rise-brazil 23/02/2024 Por Mariana Sebastião (Cidacs/Fiocruz Bahia) A taxa de suicídio entre os jovens cresceu 6% ao ano no Brasil entre 2011 e 2022. Por sua vez, as taxas de notificação para autolesões na faixa etária de 10 a 24 anos aumentaram 29% no mesmo período. O número foi… Leia mais: Estudo mostra taxas de suicídio e autolesão entre jovens em alta no Brasil
Fonte: https://portal.fiocruz.br/en/news/2024/02/study-shows-suicide-and-self-injury-rates-rise-brazil 23/02/2024 Por Mariana Sebastião (Cidacs/Fiocruz Bahia) A taxa de suicídio entre os jovens cresceu 6% ao ano no Brasil entre 2011 e 2022. Por sua vez, as taxas de notificação para autolesões na faixa etária de 10 a 24 anos aumentaram 29% no mesmo período. O número foi… Leia mais: Estudo mostra taxas de suicídio e autolesão entre jovens em alta no Brasil

